Manual¶
Demultiplexing requires two count matrices (variant-by-cell) of reads or UMIs
for each variant in each cell: AD for alternative allele and DP depth
(i.e., summary of alternative and reference alleles). These two matrices can be
obtained by genotyping a list of variants in each cell. We provide a guideline
for cellular genotyping with a recommendation of cellSNP that is developed by
us, too.
Once the genotypes for each cell have been obtained, e.g., in VCF format, or two
sparse matrices AD and DP, we can apply Vireo for demultiplexing.
Demultiplexing single cells¶
By default, Vireo works without any known genotype information for pooled samples. However, if any of genotype of these samples are known or can be obtained, e.g., by bulk RNA-seq, exome-seq, it is still useful to add them, not only allowing us to align the deconvoluted samples to its identity, but also can benefits the doublets identification, especially if the coverage or the loaded cells per sample is low.
Depending the availability of genotype information, we provide four strategies to demultiplex scRNA-seq data.
without any genotype:
vireo -c $CELL_DATA -N $n_donor -o $OUT_DIR
with genotype for all samples (genoTag: GT, GP, or PL; default is PL, please choose the existing one)
vireo -c $CELL_DATA -d $DONOR_GT_FILE -o $OUT_DIR
Optionally, -N can be provided if it is samller than that in DONOR_GT_FILE for finding the relevant subset of donors.
Note For efficient loading of donor VCF file, we recommend subset it
bcftools view donor.vcf.gz -R cellSNP.cells.vcf.gz -Oz -o sub.vcf.gzAlso, add-sor-Sfor subsetting samples.with genotype for part of the samples (n_donor is larger than that in DONOR_GT_FILE)
vireo -c $CELL_DATA -d $DONOR_GT_FILE -o $OUT_DIR -N $n_donor
with genotype but not confident (or only for subset of SNPs)
vireo -c $CELL_DATA -d $DONOR_GT_FILE -o $OUT_DIR --forceLearnGT
Viroe supports the cell data in three formats:
- a cellSNP output folder containing VCF for variants info and sparse matrices
ADandDP(recommended) - standard VCF file with variants by cells
- Vartrix outputs with three files: alt.mtx,ref.mtx,barcodes.tsv
Vireo full arguments¶
Type vireo -h for details of all arguments:
Usage: vireo [options]
Options:
-h, --help show this help message and exit
-c CELL_DATA, --cellData=CELL_DATA
The cell genotype file in VCF format or cellSNP folder
with sparse matrices.
-N N_DONOR, --nDonor=N_DONOR
Number of donors to demultiplex; can be larger than
provided in donor_file
-o OUT_DIR, --outDir=OUT_DIR
Dirtectory for output files [default:
$cellFilePath/vireo]
Optional input files:
--vartrixData=VARTRIX_DATA
The cell genotype files in vartrix outputs (three
files, comma separated): alt.mtx,ref.mtx,barcodes.tsv.
This will suppress cellData argument.
-d DONOR_FILE, --donorFile=DONOR_FILE
The donor genotype file in VCF format. Please filter
the sample and region with bcftools -s and -R first!
-t GENO_TAG, --genoTag=GENO_TAG
The tag for donor genotype: GT, GP, PL [default: PL]
Optional arguments:
--noDoublet If use, not checking doublets.
-M N_INIT, --nInit=N_INIT
Number of random initializations, when GT needs to
learn [default: 50]
--extraDonor=N_EXTRA_DONOR
Number of extra donor in pre-cluster, when GT needs to
learn [default: 0]
--extraDonorMode=EXTRA_DONOR_MODE
Method for searching from extra donors. size: n_cell
per donor; distance: GT distance between donors
[default: distance]
--forceLearnGT If use, treat donor GT as prior only.
--ASEmode If use, turn on SNP specific allelic ratio.
--noPlot If use, turn off plotting GT distance.
--randSeed=RAND_SEED
Seed for random initialization [default: none]
Discriminatory variants¶
Given a set of variants for which estimated genotypes are available, the Vireo software implements a heuristic to define a minimal and informative set of discriminatory variants. This set of variants can be used to perform qPCR-based genotyping or for other targeted genoytping methods. Briefly, this algorithm prioritises variants with largest information gain in splitting samples.
For any donor genotype file in VCF format, especially the output from Vireo,
GT_donors.vireo.vcf.gz, the GTbarcode function can be used to generate
the minimal set of discriminatory variants by the following command line:
GTbarcode -i $dir/GT_donors.vireo.vcf.gz -o $dir/GT_barcodes.tsv --randSeed 1
By default, this function filters out variants with <20 UMIs or >0.05 reads
aligned other alleles except the annotated reference and alternative alleles.
In case the variants with homozygous alternative alleles are not wanted, the
arguments --noHomoAlt can be used. By default, this GTbarcode function
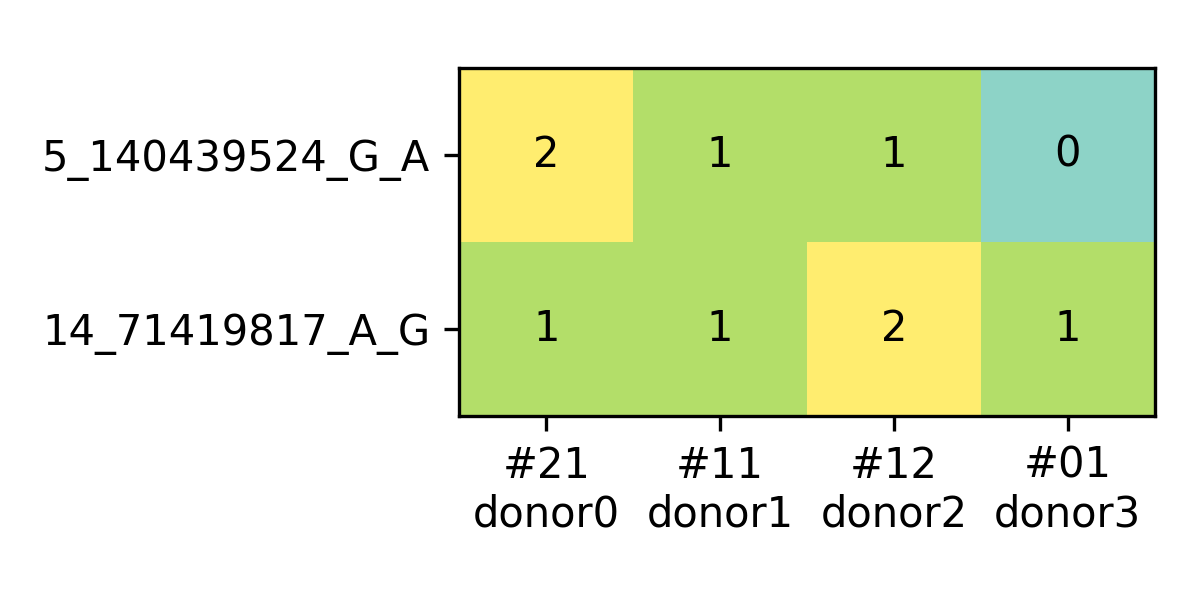
will also generate a figure for the identified genotype barcode, as following
(based on example data in the repo),
vireoSNP module usage¶
Besides the command line usage for designed donor deconvolution, we also provide tutorials on the usage of vireoSNP as a standard Python module for both donor deconvolution and general cell clustering based on allelic ratio: vireoSNP_usage.ipynb
Example data¶
In order to test vireo and illustrate the usage, we provide a test data set, also some demo scripts.
This example data set contains 952 cells from 4 samples. The genotypes for these four samples are also provided.